|
Expert Views :: An "Array" of Possibilities for Wildlife Caren Helbing, Ph.D. Fish, frogs, marine mammals, birds, alligators, and other wildlife all serve as indicators of the presence of endocrine disruptors (EDs) in the "real" world. Changes in sex ratios and sexual characteristics and alterations in hormone levels, growth, and mortality have long been used in wildlife as whole-organism measures to assess the impact of these chemicals. Although this information is useful, these measures provide limited information regarding the molecular mechanisms of action, which are critical for risk assessment and the prediction of detrimental effects within and across species at the organism and population level. Exposure to an ED or mixture of chemicals has the effect of mimicking, inhibiting, or altering normal hormonal pathways. Despite the many different effects that hormones have, a common denominator is that hormonal signals change the way target cells function by altering gene expression and/or protein function. Recent advances in the area of genomics have provided a new opportunity for combining the extensive information on whole-organism measures with novel molecular endpoints that can give insight into mechanisms of action. Genomics is a very broad term that includes identification of genes and gene products and the study of the genomic sequence, gene expression, and protein profiling. The collection of genes in a cell, called a genome, is shared by all cells in the body and constitutes the genetic "blueprint" of living organisms. The genome resides in the nucleus of the cell and contains all the information necessary for normal protein synthesis outside the nucleus. Because the genome and the sites of protein synthesis are physically separated, an intermediary messenger is required to relay the important information from the genome to the protein synthesis machinery. This messenger makes an exact copy of the gene's DNA (deoxyribonucleic acid) by transcribing the information into mRNA (messenger ribonucleic acid), which can shuttle between cellular compartments. The mRNA associates with protein synthesis machinery, which then produce the specified protein by interpreting the genetic code. The collection of mRNA transcripts inside a cell is called a "transcriptome." Transcriptomes give us some idea about what proteins may be produced in the cell. The two most common ways to look at transcriptome components are DNA arrays and quantitative real-time polymerase chain reaction (QPCR). Using DNA arrays, a large number of genes can be simultaneously monitored for their expression levels in a given target tissue. DNA sequences of interest are precisely spotted in an arrayed pattern and immobilized on a nylon or glass substrate. Then, the transcriptome is extracted from the tissue of interest, fluorescence labeled or radio labeled, and allowed to hybridize with the immobilized DNA. Differences in the expression of specific genes are then determined by the degree of label observed on each spot compared to the controls. The interrelationships between the expression levels of different gene transcripts can reveal specific "signatures" that reflect a particular mode of action. This particular approach has been used to show that changes in gene expression occur upon exposure to acetochlor and thyroid hormones in frogs (1, 2) and estrogens in fish (3, 4). 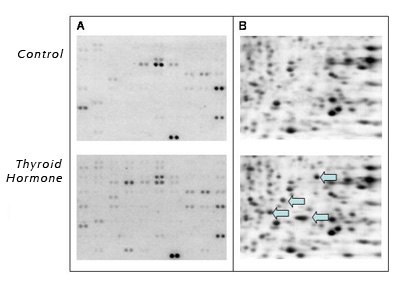 Comparison of (A) DNA array and (B) two dimensional SDS-PAGE analysis of proteins from bullfrog (Rana catesbeiana) tail fins two days after injection with vehicle control or thyroid hormone. The arrows indicate some proteins whose levels increased in response to thyroid hormone treatment. Figure 1A shows examples of different profiles from the tail fin of Rana catesbeiana bullfrog tadpoles comparing animals exposed to thyroid hormones for two days with untreated controls. Only days later are the physical effects of thyroid hormones clearly apparent with regression of the tail as part of a precociously induced tadpole metamorphosis! Since gene expression changes often precede morphological and physiological effects, discerning gene expression profiles can be used as markers for specific chemical mechanisms of action over a much shorter time frame. Although gene profiling using arrays is very powerful because it can identify interrelationships between gene transcripts, low abundance transcripts present a challenge for reliable detection and the results are, at best, semi-quantitative. Thus coupling this technology with the more sensitive QPCR technique for particular target genes is essential. QPCR is far more reliable for low-copy number transcripts and truly is quantitative. It can be performed on minute quantities of mRNA as starting material and is highly conducive to replicate samples such as tissue biopsies (5, 6). However, it is limited in the number of gene transcripts that can be examined at a time. A completely different approach is taken for proteins. Proteins are the major "workers" of the cell and function as enzymes, structural components, and adaptor molecules. Some proteins are common to all cells, whereas others are specific to only certain types of cells. The entire complement of proteins inside a cell is called - you guessed it a proteome. The proteome includes post-translational modifications such as phosphorylation that change the activities of proteins (like on and off switches). A common way to analyze proteins is by using two dimensional polyacrylamide electrophoresis (2D-SDS-PAGE) followed by mass spectrometric analysis to identify proteins of interest. Patterns of spots are compared to each other to identify the appearance or disappearance of protein spots (Figure 1B). Often, the results comparing the transcriptome and the proteome to each other show little resemblance (2). This is actually a good thing in that it provides for a much broader mechanistic sweep of potential biomarker identification. Transcriptomic and proteomic analyses produce a LOT of data per experiment and require very intensive use of bioinformatics (the use of computers to manage biological data) for appropriate statistical analysis, database comparison, and protein fragment identification. And that's just for a limited set of experiments. Multiply that by several wildlife species and target tissues and the complexity of analysis becomes huge indeed! One of the big challenges that currently face researchers and regulators is managing and consolidating the data into a usable, retrievable format. The easiest way to examine a transcriptome or proteome is to have the genome of the species of interest sequenced. This information is used to design DNA arrays and to determine protein identities by mass spectrometry. Unfortunately, very few of the genomes available come from wildlife species and efforts to enhance our genomic database on wildlife species are vital. Through initiatives such as the Tree of Life (National Science Foundation), representatives from a broad range of phyla should be sequenced soon. But what impact does this have on the available tools for use on wildlife species? These initiatives will certainly be helpful, but creativity for those interested in studying wildlife is also required. The differences in genetic sequences between even closely related species can be quite divergent, and this can be problematic in hybridization-based experiments involving DNA arrays. However, careful array design can broaden the utility of these arrays to species other than the source of the genetic information. For example, the arrays in Figure 1 were designed with primarily Xenopus laevis sequences, but were successfully used on Rana catesbeiana samples. This kind of heterologous hybridization approach could yield vital information from species that have important ecological relevance but limited or no genetic information. Once interesting genes are identified, they can be selectively cloned from the species of interest for QPCR-based analysis. Genomics-based tools for wildlife have many challenges due to the diversity of wildlife they are meant to be used on. However, the challenges are worth tackling since wildlife hold important keys to human environmental health. References
|